Permission to Manufacture Class A-B Medical Devices in India - Form MD 3 & MD 5

The application for manufacturing, sale or distribution of any medical device includes a tedious list of processes and forms CliniExperts team consists of experts working in the same domain with practical experience and knowledge of the governing rules. Hence, we provide a one-stop solution for all the licensing and approval needs of your firm.
Permission to Manufacture Class A-B Medical Devices – Overview
Manufacturing medical devices requires stringent processes to be completed under the laid down rules and regulations by Central Drug Standard Control Organisation (CDSCO). In addition, applicants who intend to manufacture Class A or Class B medical devices have to go through a procedure by applying for a license to sell or distribute medical devices. The CDSCO ensures quality and safety of medical devices sold in the market in this manner.
Such applicants have to apply to the State Licensing Authority for approval from the Ministry of Health and Family Welfare's online portal, depending on the applicant's location. An application has to be submitted in FORM MD-3 along with necessary documents for procuring a license as FORM MD-5.

Who Can Apply?
A manufacturer can apply for Permission to manufacture Class A or Class B Medical Devices via FORM MD-3 and FORM MD-5.
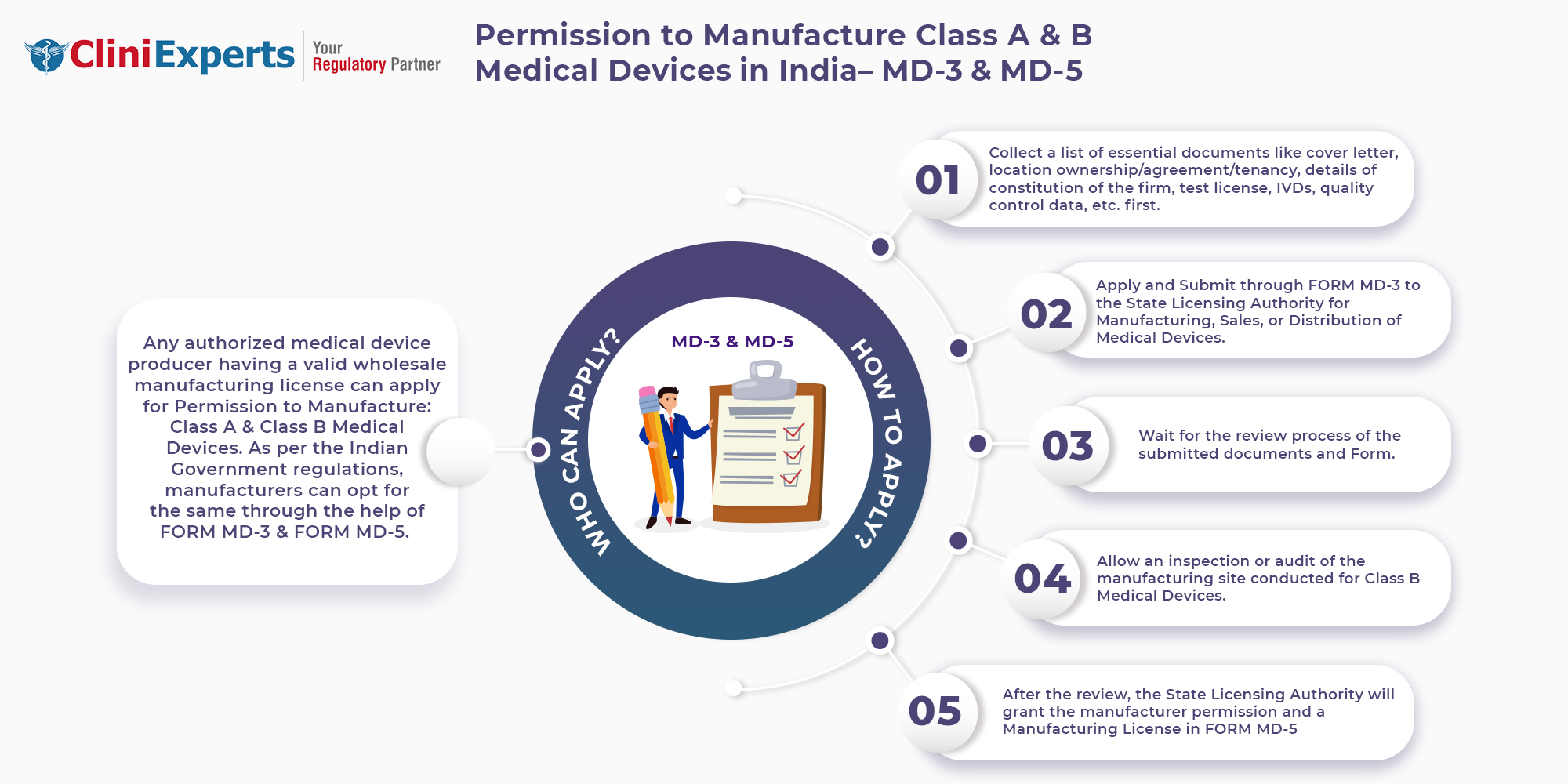
How To Apply?
The Applicant must follow the following process:
-

Step 1: Collect a list of essential documents like Cover letter, Location ownership/ agreement/ tenancy, details of constitution of the firm, test license, IVD's, quality control data
-

Step 2: Apply and submit through Form MD-3 to the State Licensing Authority for manufacturing, sales, or distribution of Medical Devices.
-

Step 3: Wait for the review process of the submitted documents and Forms
-

Step 4: Allow an inspection of the manufacturing site conducted for Class B Medical Devices
-

Step 5: After the review, the State Licensing Authority will grant the manufacturer permission and the manufacturing license in Form MD-5

Validity
A license obtained by approval as FORM MD-5 is valid permanently, as long as the license retention fee is paid timely, before completion of five years from the date of issuing by the State Licensing Authority as per the Second Schedule. However, a License can be suspended or cancelled by the State Licensing Authority based on competent reasons at any point of time.

Fee Involved
For manufacturing medical devices, a licensing fee is required for the application which includes:- One site manufacturing fee for Class A or Class B medical devices is 5000 INR.
- A distinct fee for each medical device from Class A or Class B consists of 500 INR (per medical device)
Important Documents

The required documents for the application stated by the CDSCO includes:
- Device Master File
- Site Master File
- Quality Management System Documents
Timeline to get
MD 5
from State Licensing Authority
4 to 5
MONTHSEssential Tips
- Applicant should make sure that all the requirements are met for the manufacturing site for Class B medical devices, under the conditions of Quality Management System as stated in the Fifth Schedule.
- Applicant should take care of the formatting of Device Master Files and the Site Master File as per the format under the Medical Devices Rules, 2017.
- The technical documents for approval should conform with the Medical Devices Rules, 2017.
Expert Advise
Quality control data submitted must be generated based on a validated test while applying for the license.
Related Services
Frequently Asked Questions
Will the manufacturer have an option to choose Notified body?
The Notified body accredited under sub-rule (1) of Rule 13 shall be competent to carry out an audit of manufacturing sites of Class A and Class B medical devices to verify their conformance with the Quality Management System and other applicable standards as specified under these rules in respect of such medical devices as and when so advised by the State Licensing Authority.

If a manufacturing firm is complying with ISO/IEC standards, would it still need to follow BIS standards?
- The medical device shall conform to the standards laid down by the Bureau of Indian Standards established under section 3 of the Bureau of Indian Standards Act, 1985 (63 of 1985) or as may be notified by the Ministry of Health and Family Welfare in the Central Government, from time to time.
- Where no relevant standard of any medical device has been laid down under subrule (i), such device shall conform to the standard laid down by the International Organization for Standardization (ISO) or the International Electro Technical Commission (IEC), or by any other pharmacopoeial standards.
- In case of the standards which have not been specified under sub-rule (i) and sub-rule (ii), the device shall conform to the validated manufacturer’s standards.

Does the requirement in the fourth schedule which specifies that the manufacturers have to submit an undertaking that they comply with the provisions of the fifth schedule applicable to application for license to manufacture?
Undertaking signed stating that the manufacturing site is in compliance with the provisions of the Fifth Schedule of Medical Device Rules 2017needs to be submitted in case of manufacture of Class B, C and D medical devices.
Will business continuity be considered for devices already in market, but not yet notified, if they are brought under the list of notified devices?
Once devices are brought under notified categories, the manufacturer has to comply with Medical Device Rules, 2017.
For devices, already in market and notified later, would the requirement of local clinical investigation/evaluation be waived off?
The medical device on the basis of their intended use will be deliberated on case to case basis & data available, to substantiate their safety and effectiveness. The matter may also be placed before SEC.