Permission to manufacture/import new medical device in India - Form MD 26 & Form 27

Get experts assistance to avail Permission to manufacture/import new medical device which does not have its predicate device in India -As per MDR 2017
Permission To Manufacture/Import New Medical Device (MD 26, 27) – Overview

Who Can Apply?
To get an import/manufacturing license for the sale/distribution of a medical device that does not have a predicate medical device, the importer/manufacturer must make an application to the Central Licensing Authority. The import license applicant must do this via an authorized agent having a manufacturing or wholesale license for the sale or distribution of medical devices. The application should be made to the Central Licensing Authority via an online portal of the Ministry of Health and Family Welfare in the Central Government using Form MD-26 and the permission is received in the Form MD-27.
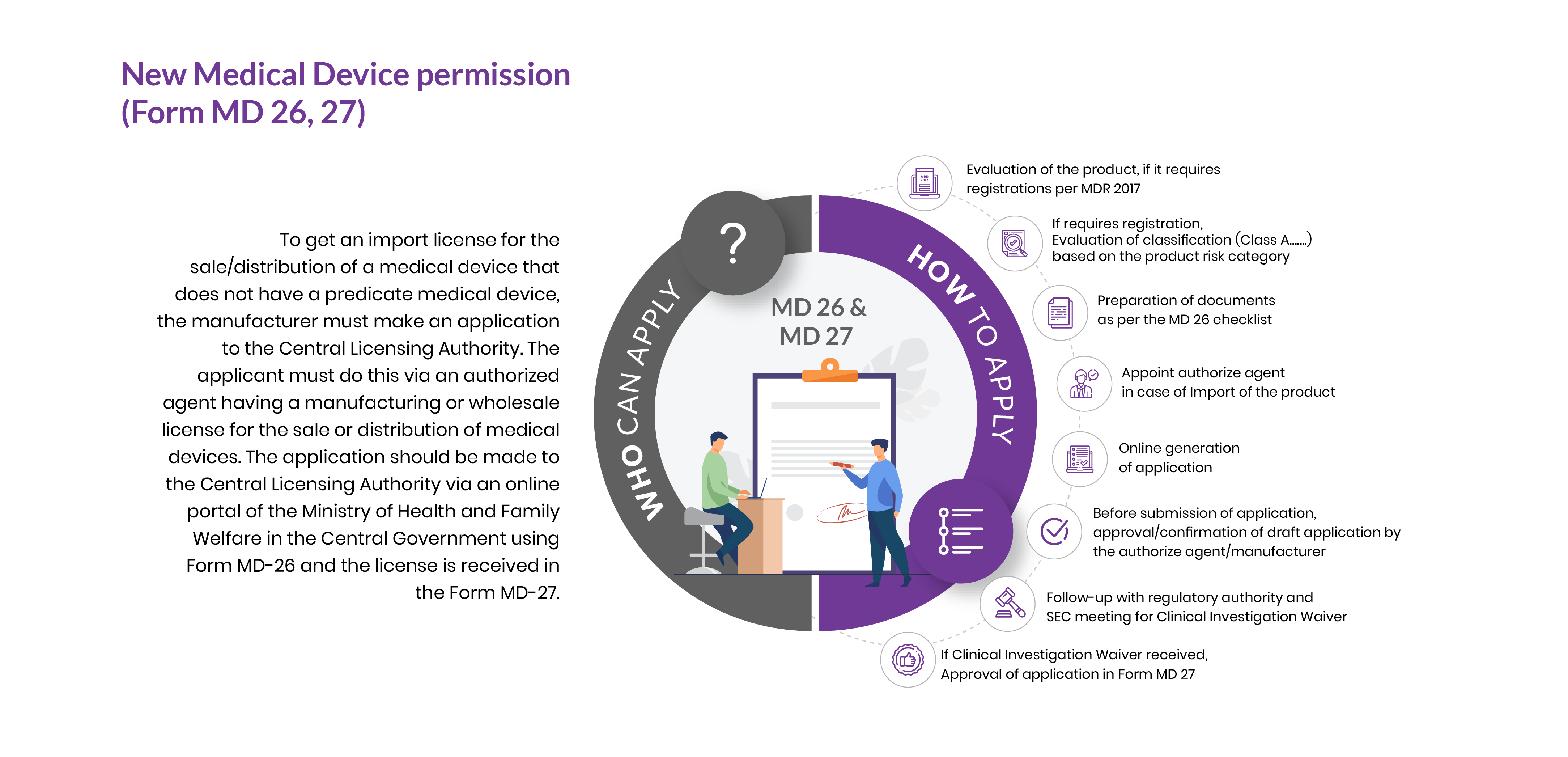
How To Apply?
The Applicant must follow the following process:
-

Evaluation of the product, if it requires registrations as per MDR 2017
-

If requires registration, Evaluation of classification (Class A, B, C, & D) based on the product risk category
-

Preparation of documents as per the MD 26 checklist
-

Appoint authorized agent in case of Import of the product
-

Online generation of application
-
Before submission of application, approval/confirmation of draft application by the authorised agent/manufacturer
-
Follow-up with regulatory authority and SEC meeting for Clinical Investigation Waiver
-
If Clinical Investigation Waiver received, Approval of application in Form MD 27

Validity
As per MDR 2017 the validity of the license is not specified as of now.

Fee Involved
INR 50000 is the prescribed fee for taking the permission to manufacture/import new medical device that does not have its predicate device.
Important Documents

- Wholesale License
- A copy of a foreign manufacturing plant or registration of establishment. This copy must be notarized.
- Free Sale Certificate from GHTF
- Data for Design Analysis
- Bio-compatibility tests data
- Risk management data
- Animal performance data
- Safety and performance data
- Pharmacovigilance data
- A copy of a letter which shows approval status in countries like the United Kingdom, United States of America, Australia, Canada, Japan, EU. The approval letter must contain the number and date
- All the specific details of the country where this device has been sold for the past two years
Timeline to get
Form 27
from CDSCO
2 to 3
MONTHSEssential Tips
The applicant looking for permission to import medical devices must ensure these essentials are followed.
- An essential requirement is an appointment with an Authorized agent. An authorized agent is a person having a license to manufacture a medical device for sale/distribution or a wholesale licenses for the sale or distribution of medical devices.
- The applicant must check for approval in Global Harmonization Task Force (GHTF) countries like USFDA, Australia, Canada, Japan, and the EU.
- The investigational medical device must be approved in GHTF countries and sold for at least two years in that country.
- The applicant must also check whether a waiver from clinical trials is applicable.
- To get a waiver from clinical trials, the investigational medical device must comply with other conditions specified in MDR 2017.
- If the authority has not waived off the clinical trials of the investigational medical device, the applicant must fill FORM MD-22 to obtain permission for conduction clinical trials.
- The applicant must provide all the necessary information required by the Central Licensing Authority within 90 days from the date of intimation.
Expert Advise
The medical device classification must be done as per the rules mentioned in MDR 2017.
To get a waiver from clinical trials by the CDSCO, the approval of the medical device in GHTF countries and other conditions must be checked.
If satisfactory data is submitted by the manufacturer of the investigational medical device in GHTF countries, where this product has been marketed for at least two years, the manufacturer do not need to fill Form MD-22, hence get a waiver from the clinical trial.
Related Services
Frequently Asked Questions
Which classification will be considered if the medical device classification is different in the GHTF countries and in India?
If the medical device classification is different in India and GHTF countries, the higher class will be considered.

Who will be responsible for the Post Marketing Surveillance (PMS)?
The authorized agent/Manufacturer who has obtained an import license/Manufacturing License is responsible for the PMS.
If the first importer has obtained the approval for new investigational medical device, do the subsequent applicants also need to get an approval? Can they just apply for the import license?
If the first importer has obtained the approval for new investigational medical device, then subsequent applicant can just apply for the import license of the same medical device.
Will the clinical investigation be waived off if a CE marking is available when importing a medical device that does not have a predicate device?
The clinical investigation in India may be waived off if the medical device has been approved by regulatory authorities and marketed in UK, USA, Canada, or Japan for at least two years; if the data of safety, performance, and pharmacovigilance submitted to the Central Licensing Authority is satisfactory.
When to submit the Periodic Safety Update Report (PSUR)?
The applicant who has been granted permission via Form MD-27 must submit the PSUR from the date of launch of the device in the market followed by submission every six months for the first two years. Later, the PSUR must be submitted annually for two more consecutive years. The PSUR should be submitted to the Central Licensing Authority.